 联系我们
联系我们摘要:USP18介导的去ISG化(deISGylation)建立的动态平衡对结核病的发生、发展和转归有重要影响。
干扰素诱导基因15 (interferon-stimulated gene 15,isg15)的表达受Ⅰ型干扰素诱导,该基因编码的蛋白ISG15可以分别通过E1、E2和E3酶的作用共价修饰靶蛋白,此过程被称为ISG化(ISGylation)。宿主蛋白的ISG化广泛参与天然免疫例如宿主的抗病毒过程。泛素特异性蛋白酶18 (ubiquitin-specific protease 18,USP18)作为一种去泛素化酶(deubiquitinase,DUB)可以去除靶蛋白偶联的 ISG15,并通过抑制Ⅰ型干扰素信号通路来抑制宿主的免疫应答。

泛素特异性蛋白酶18 (ubiquitin-specific protease 18 ,USP18)和干扰素诱导基因15 (interferonstimulated gene 15,isg15)介导的ISG化修饰是机体参与抗病原微生物感染的重要组分。越来越多的研究发现,靶向ISG15和USP18及其底物的蛋白质翻译后修饰有望开发成为新型抗感染治疗的策略之一。
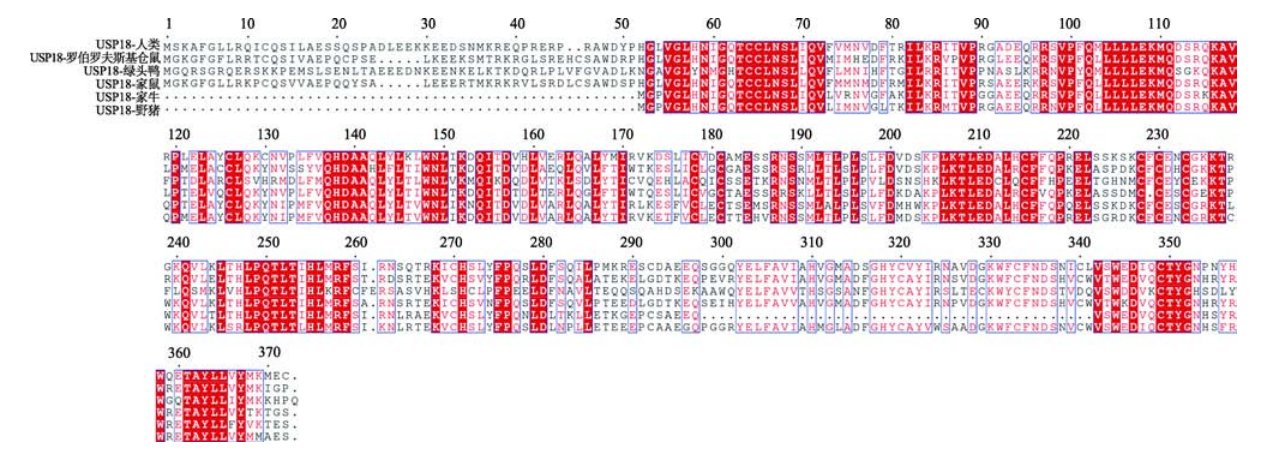
USP18是一个大小约 43 kDa的特异性蛋白酶,ISG15和USP18均可被干扰素(interferon,IFN)诱导。通过比较人类(Homo sapiens)、家鼠(Mus musculus)、家牛(Bos taurus)、野猪(Sus scrofa)、罗伯罗夫斯基仓鼠(Phodopus roborovskii)、绿头鸭(Anas platyrhynchos)等物种中USP18的氨基酸序列,发现不同物种中的 USP18 具有高度的同源性。USP18在不同物种中都含有高度保守的序列,该保守序列包括半胱氨酸残基和组氨酸残基位于 USP18 的活性中心,这也是 USP家族蛋白酶所特有的结构。
ISG15 能够参与并激活多种信号通路,发挥抗病毒免疫的功能,与之相反,USP18会限制NF-κB、JNK和NFAT等通路的激活,负调节炎症反应,调节T淋巴细胞和辅助性T细胞的活化。USP18在Ⅰ型IFN中的作用下依赖于α/β干扰素受体2 (interferon alpha/beta receptor 2,IFNAR2),并负调控Ⅰ型IFN信号通路。USP18可以切割ISG15与靶蛋白之间的异肽键,还
能在结合后立即切割ISG15的LRLRGG序列,在由ISG15前体加工为成熟的ISG15过程中发挥重要作用。鉴于ISG15和USP18在介导宿主蛋白泛素化和去泛素化以及免疫应答中的重要作用,来自西南大学生命科学学院现代生物医药研究所的研究团队对ISG15和USP18及其突变体在结核分枝杆菌(Mycobacterium tuberculosis,Mtb)等感染免疫应答中的作用机制进行解读,并探讨了其作为治疗靶点的潜力。
ISG15特异性蛋白酶——USP18
蛋白去泛素化是由一组去泛素酶(deubiquitinase,DUB)介导的泛素化的反向过程。DUB家族有90多个成员。泛素特异性蛋白酶USP18和USP20属于DUBs的USP亚家族,并介导靶蛋白的去泛素化。USP20靶向多种蛋白质底物,包括HIF1α、β-肾上腺素能受体、TNF受体关联因子6(TNF receptor associated factor 6,TRAF6)和Claspin,通过去泛素化调节Toll样受体4(toll-like receptor 4,TLR4)信号转导和DNA损伤修复。USP18最初被鉴定为DUB,后来被发现也具有去ISG化酶活性,因为小鼠中USP18缺失或非活性突变体usp18C61A会导致高水平的ISG化。此外,USP18在ISG15前体加工产生成熟的 ISG15 分子过程中也发挥作用,但在 USP18缺陷小鼠中,ISG15前体也能被加工成其成熟形式,重组ISG15前体可以被II型肺泡上皮细胞A549的一种大小100 kDa的酶正确加工,且该酶是酵母泛素特异性肽酶 1(ubiquitin-specific protease,USP1)同源物,活性不受I型IFN刺激的影响。这提示除USP18 之外,还有其他 ISG15 特异性蛋白酶。
一些E2和E3酶在ISG15化和泛素化过程中的功能具有重叠,也意味着存在可以作为ISG15 特异性蛋白酶的多功能 DUB,包括 USP2、USP5、USP13 和 USP14 在内的几种 DUB 都被认为是ISG15 特异性蛋白酶的候选物。但是,小鼠中USP18基因的缺失会导致组织中ISG15 结合物大量增加,而不会影响泛素结合物的水平,这表明USP18是ISG15特异性蛋白酶,可将ISG15从靶蛋白上去除。
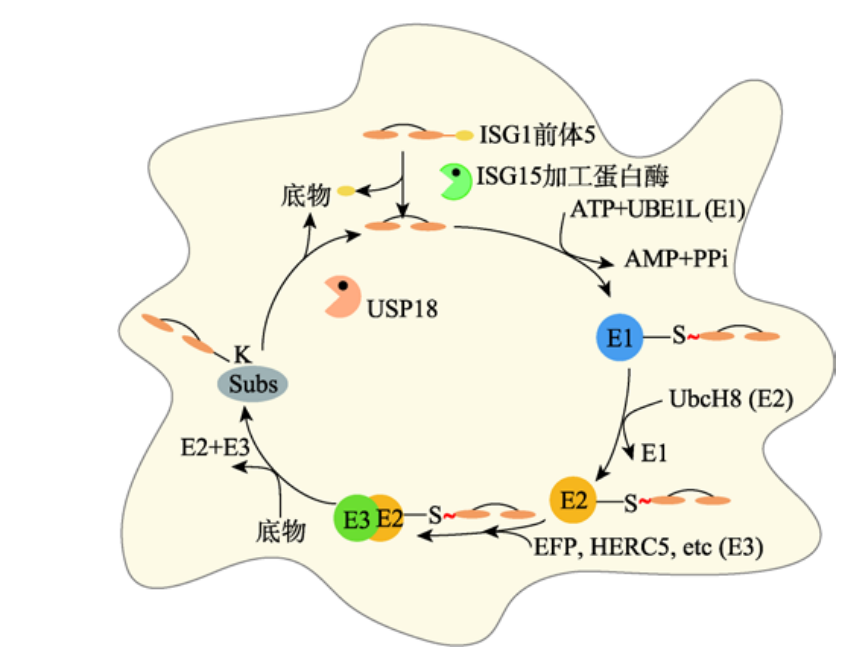
ISG15前体经ISG15加工酶变成ISG15,分别与E1、E2、E3泛素酶结合,将ISG15结合在靶蛋白上,USP18能够从靶蛋白上去除ISG15,发挥去ISG化功能。
USP18的表达主要受I型IFN诱导,而这种诱导需要通过JAK/STAT信号通路的作用。干扰素
β (interferon-beta,IFN-β)比干扰素 α (interferon-alpha,IFN-α)和双链 RNA (double strand RNA,dsRNA)诱导USP18的作用更强,但干扰素γ (interferon-gamma,IFN-γ)几乎没有诱导作用。USP18也可被脂多糖(lipopolysaccharide,LPS)诱导。干扰素调节因子2(interferon regulatory factor 2,IRF2)和干扰素调节因子3 (interferon regulatory factor 3,IRF3)都是LPS应答所必需的,LPS通过IRF3上调USP18,而IRF2可将其降至基础水平。在各种造血细胞系中,单核
细胞和巨噬细胞系能够高水平表达USP18。Skp2 (S-phase kinase associated protein 2)是S期激酶相关蛋白,属于F-box蛋白家族,在蛋白泛素化降解过程中可作为 Skp1-Cul1-F-box(SCF)蛋白复合物的重要成分识别底物蛋白,通过降解细胞周期调节蛋白而调控细胞周期。USP18 是Skp2的底物,Skp2促进USP18的泛素化和随后的蛋白酶体降解。这表明SCF-Skp2可能通过控制USP18的稳定性,调节I型IFN信号转导。然而,有大量基因例如急性髓系白血病(acute myeloid leukemia,AML)融合基因AML1-ETO诱导USP18上调的机制尚待研究。
USP18负调节先天免疫反应
ISG15和USP18在宿主对病毒感染的反应中起着重要作用。在I型IFN处理后,与野生型细胞相比,usp18C61A细胞对B型流感病毒感染有更强的抵抗力,而病毒在isg15缺失的 usp18C61A 细胞中可完全恢复复制能力。除了其酶功能外,USP18还负调控I型IFN信号转导,与Janus激酶(janus kinase,JAK)竞争结合IFNAR2。因此,与野生型细胞相比,用I型IFN 处理usp18−/−细胞导致信号转导转录激活因子 (signal transducers and activators of transcription 1,STAT1)磷酸化增加且持续时间延长,增强ISG化,并促进细胞凋亡。这与抑制USP18可增强HepG2.2.15细胞中IFN-α 的抗乙型肝炎病毒(hepatitis B virus,HBV)活性这一结论相吻合。
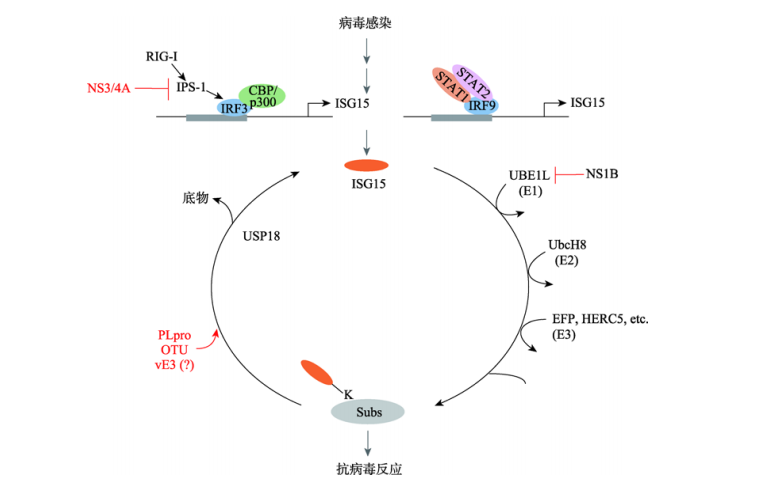
模式识别受体如RIG-I在先天免疫细胞中表达,招募接头分子IPS-1,进而触发MAVS-TBK1-IRF3 通路等信号通路的激活,ISG15能够靶向并参与修饰一系列重要的抗病毒蛋白,如STAT1、IRF3、RIG-I等,病毒感染过程中ISG15介导的ISGylation以及与底物的结合能够使宿主发挥抗病毒反应,同时USP18将负调控某些抗病毒蛋白的表达。
USP18先天缺失与相关疾病
USP18基因的缺失使细胞内的IFN水平失调,一方面可能会导致个体出现先天致死,但另一方面也能够增强癌症患者或病毒感染患者的免疫应答,促进癌症治疗和病毒清除。
ISG15缺失的个体易患孟德尔遗传易感性的分枝杆菌病(mendelian susceptibility to mycobacterial disease,MSMD),这是因为缺失ISG15会触发T细胞和自然杀伤细胞(natural killer cell,NK)产生过量的IFN-γ。USP18小鼠更容易感染Mtb,肺和脾脏中的细菌数量增加,
炎性细胞因子升高,肺部病变更严重。
蛋白质泛素化修饰是感染免疫的重点研究对象之一,去泛素酶USP18是维持细胞中ISG15共价修饰蛋白质的动态平衡和功能的关键,在DNA/RNA病毒感染期间调节病毒复制、聚集以及对宿主的易感性。HBV等病原可以逃避宿主模式识别受体的识别、抑制下游信号转导,调控宿主 USP18 等去泛素化酶活性而干扰IFN信号转导,最后逃避免疫清除。
因此针对DUBs尤其是USP18在患者中的异常表达及其调控因子研发新的靶向药物,将有助于开发新的抗感染工具。
参考文献
- Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem, 2009, 78: 363–397.
- Yasunaga J, Lin FC, Lu XB, Jeang KT. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-kappaB signaling. J Virol, 2011, 85(13): 6212–6219.
- Huijser E, Bodewes ILA, Lourens MS, van HeldenMeeuwsen CG, van den Bosch TPP, Grashof DGB, van de Werken HJG, Lopes AP, van Roon JAG, van Daele PLA,Brkic Z, Dik WA, Versnel MA. Hyperresponsive cytosolic DNA-sensing pathway in monocytes from primary Sjögren's syndrome. Rheumatology (Oxford), 2022, 61(8): 3491–3496.
- Anne-Boland-Auge, Deleuze JF, El-Chehadeh S, Anheim M, de Saint-Martin A, Pellegrini S, Marsh JA, Crow YJ, El-Daher MT. Type I interferonopathy due to a homozygous loss-of-inhibitory function mutation in STAT2. J Clin Immunol, 2023, 43(4): 808–818.
- Qi’ao Zhang, Zilu Wang, Peibo Li, Jianping Xie.USP18-mediated protein deISGylation and its role in tuberculosis and other infectious diseases.Hereditas (Beijing) 2023, 45(11): 998―1006.









